进化速率
定义与基本概念编辑本段
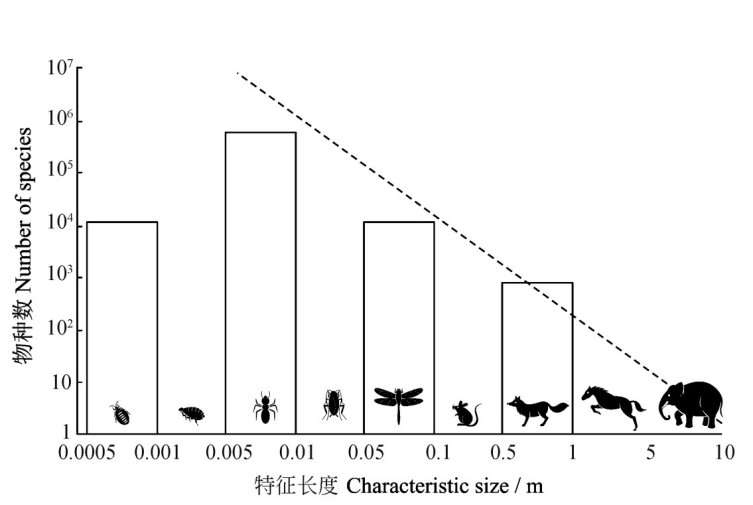
进化速率(evolutionary rate)是进化生物学中衡量生物进化快慢的关键指标。在分子层次上,通常指单位时间内核苷酸或氨基酸替换发生的频率,常用每个位点每年(或每百万年)的替换数表示。在形态学或表型层次上,则指性状状态变化的速率,如体长、颅骨特征等连续性状的进化速率。进化速率的研究可追溯至19世纪末对化石形态变化率的探讨,而20世纪60年代随着分子生物学兴起,分子进化速率成为核心研究领域。
分子进化速率的类型与度量编辑本段
分子进化速率主要分为非同义替换率(dN)和同义替换率(dS)。非同义替换涉及氨基酸改变,受选择约束较强;同义替换基本不受选择影响,可反映中性突变背景速率。常用比值为dN/dS(ω),用以判断选择压力:ω<1指示纯化选择,ω=1指示中性进化,ω>1指示正选择。此外,根据序列类型,还可区分编码区与非编码区(如内含子、基因间区)的进化速率。度量分子进化速率需依赖系统发育树和化石标定或分子钟模型,常用方法包括最大似然法和贝叶斯推断。
影响进化速率的因素编辑本段
影响进化速率的因素众多,可分为内在和外在两类。内在因素包括:世代时间(即一个世代所需时间),世代越短,单位时间内的生殖和突变累积机会越多,通常导致更快的分子进化速率,此即“世代时间效应”;生殖方式,无性生殖或自交物种往往比异交物种积累更多突变;基因组大小和结构,如基因密度、复制偏好等。外在因素主要包括环境条件,如温度、辐射水平可影响突变率;种群大小,小种群因遗传漂变增强而固定更多中性或轻微有害突变;选择强度,生态位分化或性选择等可加速特定性状的进化。此外,基因自身功能约束也至关重要:管家基因通常进化较慢,若基因具有众多互作位点或编码关键蛋白质结构域,则受较强纯化选择而速率低下。
分子钟假说及其验证编辑本段
分子钟假说由Zuckerkandl和Pauling于1962年提出,认为分子进化速率在长时间尺度上大致恒定。其基础假设为大多数分子变化是中性或近乎中性的,且突变率随时间稳定。然而,研究发现在不同支系间,分子时钟并不完全均匀,例如啮齿类相较于灵长类表现出更快的进化速率,这与世代时间效应相符。为此,发展了松弛分子钟模型(如局部分子钟、贝叶斯松弛分子钟),允许速率在不同谱系间变化,广泛应用于物种分歧时间估算。分子钟的应用极大推动了系统发育年代学,如对哺乳动物辐射时间的校正。
进化速率的生物学意义编辑本段
进化速率差异隐含了深刻的生物学信息。慢速进化的基因或区域往往行使重要功能,参与发育调控和核心代谢,如组蛋白H3和细胞色素c。快速进化的基因常与适应性相关,如免疫系统相关基因、生殖相关基因(如精液蛋白)和对环境压力响应的基因。在寄生虫与宿主、捕食者与猎物间的“军备竞赛”中,相关基因受正选择驱动而加速进化。进化速率的比较也可用于鉴定功能重要位点:例如,绝对保守的氨基酸位点通常为蛋白质结构与活性所必需。
进化速率的研究方法编辑本段
研究进化速率首先需要高质量的多序列比对和可靠的系统发育树。随后通过计算成对序列间的差异并校正多重替换(如使用Kimura双参数模型或GTR模型)来估计替代数。对于多物种比较,常用分支特异性速率估计,如利用密码子模型(如YANG、Nielsen模型)估算支系水平的dN/dS。贝叶斯方法可整合化石先验信息,同时估计系统发育、进化速率和分异时间。此外,基于形态性状的进化速率可使用似然分析,如Brownian运动模型或O-U过程。
进化速率在应用领域的贡献编辑本段
在病毒学中,进化速率的估算对于理解病毒传播动态、疫苗设计至关重要。例如,流感病毒和HIV的迅速进化需要不断更新疫苗株。在人类进化研究中,通过比较人与黑猩猩的基因组替换速率,估算出人猿分异约600万年前。癌症基因组学亦借鉴该概念:肿瘤内体细胞突变的积累速率可用于推断肿瘤演化历史和亚克隆结构。生态学中,表型进化速率可揭示物种适应辐射的驱动因素,如达尔文雀喙形的快速进化与食物资源相关。
挑战与前沿编辑本段
尽管进化速率研究已取得巨大成就,但面临的挑战不容忽视。首先,时间的刻度问题:短时间尺度下的进化速率估算常受祖先多态性、选择干扰以及测年精度限制。其次,突变率本身的变异性:除遗传因素外,表观遗传修饰(如DNA甲基化)也会影响突变率,且环境胁迫可诱导突变率升高(突变性)。最后,跨层次整合问题:如何将分子进化速率与表型进化速率统一在共同理论框架内仍是热点。近年来,随着古基因组学的进展,可从古老DNA直接估计历史进化速率;单细胞测序和谱系追踪技术开始研究多细胞生物体细胞进化速率。这些前沿将深刻重塑对进化速率本质的理解。
参考资料编辑本段
- Kimura, M. (1968). Evolutionary rate at the molecular level. Nature, 217(5129), 624-626.
- Zuckerkandl, E., & Pauling, L. (1962). Molecular disease, evolution, and genic heterogeneity. In Horizons in Biochemistry (pp. 189-225). Academic Press.
- Yang, Z. (2007). PAML 4: maximum likelihood analysis of phylogenetic trees. Molecular Biology and Evolution, 24(8), 1586-1591.
- Ohta, T. (1973). Slightly deleterious mutant substitutions in evolution. Nature, 246(5428), 96-98.
- Bromham, L., & Penny, D. (2003). The modern molecular clock. Nature Reviews Genetics, 4(3), 216-224.
- Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870-1874.
- Drummond, A. J., & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7(1), 214.
- Nei, M., & Kumar, S. (2000). Molecular Evolution and Phylogenetics. Oxford University Press.
附件列表
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。