溶酶体贮积症
溶酶体贮积症(Lysosomal storage disease, LSD)是一类由溶酶体功能缺陷引起的遗传性代谢疾病,其共同特征为未降解或部分降解的底物在溶酶体内异常蓄积,进而导致细胞损伤、器官功能障碍及进行性临床表现。目前已识别超过70种LSD,多为常染色体隐性遗传,少数为X连锁遗传(如Fabry病、Hunter综合征)。总体发病率约为1/5000~1/8000活产婴儿,但不同亚型发病率差异显著。 ADFASDFAF23RQ23R
病因与发病机制编辑本段
 ADSFAEQWER353423413434
ADSFAEQWER353423413434 溶酶体贮积症
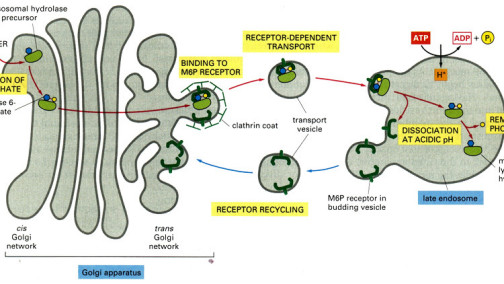
LSD的分子基础主要涉及编码溶酶体水解酶、转运蛋白、激活蛋白或保护蛋白的基因突变,导致酶活性缺失、底物转运障碍或辅因子缺失。根据贮积物质类型,LSD可大致分为:鞘脂贮积症(如戈谢病、尼曼-匹克病、Fabry病)、黏多糖贮积症(MPS I~IX型)、糖蛋白贮积症(如岩藻糖苷贮积症)、糖原贮积症Ⅱ型(庞贝病)、神经元蜡样脂褐质沉积症(NCLs)等。其共同病理特征为:溶酶体肿胀、细胞空泡化,继而触发细胞凋亡、炎症反应、自噬功能紊乱及内质网应激等次级效应,最终导致多系统损害。 ADFASDFAF23RQ23R
临床表现编辑本段
LSD的临床谱系极其广泛,从胎儿期发病的严重类型(如MPS I Hurler型)至成人期轻微变异型(如Fabry病心脏变异型)均可出现。常见表现包括:
1. 神经系统:发育迟缓、智力倒退、共济失调、癫痫、肌张力障碍、视听力丧失等(如NCLs、Krabbe病)。
2. 骨骼系统:多发性骨发育不良、关节挛缩、身材矮小(如MPS I型特征性‘Hurler容貌’)。 ADFASDFAF23RQ23R
3. 内脏器官:肝脾肿大(戈谢病、尼曼-匹克病)、心脏瓣膜病(Fabry病)、肾功能不全(Fabry病)。 ADSFAEQWER353423413434
4. 其他:面部粗陋、角膜浑浊、血管角质瘤、反复感染等。病程呈进行性,部分类型可累及胎儿(如MPSⅦ型导致的胎儿水肿)。
ADSFAEQWER353423413434
诊断编辑本段
诊断流程包括临床评估、酶活性检测、基因测序及生物标志物分析。 ADFASDFAF23RQ23R
酶活性检测:是金标准,利用人工荧光底物或放射性标记底物在白细胞、成纤维细胞或干血斑中测定特定酶活性(如葡萄糖脑苷脂酶活性检测诊断戈谢病)。 ADFASDFAF23RQ23R
基因检测:通过Sanger测序或二代测序发现致病基因突变,辅助确认携带者及产前诊断。
ADFASDFAF23RQ23R
生物标志物:如血浆壳三糖酶活性(戈谢病)、糖胺聚糖定量(MPS)、鞘脂类谱(LC-MS/MS检测多种鞘脂)等用于筛查和监测。 ADSFAEQWER353423413434
影像学(X线、MRI)可显示骨骼、脑实质等特征性改变(如MPS的‘J形蝶鞍’)。产前诊断采用羊水细胞或绒毛膜活检。 ADSFAEQWER353423413434
治疗编辑本段
治疗策略逐步个体化,主要包括:
酶替代疗法:重组人源酶定期静脉输注,用于部分非神经病变型LSD(如戈谢病、Fabry病、庞贝病、MPS I、MPS VI等),可改善内脏症状但难以跨越血脑屏障。
底物减少疗法:抑制底物生物合成,例如米格司他(Miglustat)用于戈谢病轻型,依利格司他(Eliglustat)用于CYP2D6快代谢型患者。
造血干细胞移植:适用于某些MPS、Krabbe病等早期患者,可提供持续的酶源,改善神经性症状,但存在移植相关风险。 ADFASDFAF23RQ23R
基因治疗:利用AAV载体等将功能基因导入体内,多项临床试验正在进行(如MPS III、庞贝病),展现出持久疗效潜力。 ADSFAEQWER353423413434
对症治疗:包括抗癫痫、理疗、矫形手术、呼吸支持等,提高生活质量。
研究进展与展望编辑本段
近年LSD领域进展迅速:新药研发聚焦于穿透血脑屏障的酶制剂(如鞘内注射、融合肽载体);小分子药物如底物减少疗法联合分子伴侣疗法(如Migalastat用于Fabry病特定突变型);CRISPR-Cas9编辑技术矫正突变的体外及体内研究已进入临床前阶段。此外,新型生物标志物(如Lyso-Gb3、鞘氨醇-1-磷酸)和新生儿筛查的推广(如MPS I已在多国纳入)有助于早期干预。尽管挑战犹存,LSD已成为遗传性代谢疾病转化医学的典范领域。 ADSFAEQWER353423413434
参考资料编辑本段
- Platt FM, d'Azzo A, Davidson BL, et al. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4(1):27.
- Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249-254.
- Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3(12):954-966.
- Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-L-iduronidase (laronidase). J Pediatr. 2004;144(5):581-588.
- Aerts JM, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders. J Inherit Metab Dis. 2011;34(5):961-971.
- Biffi A, De Palma E, Liso A, et al. Lentiviral vector common integration sites in preclinical models and clinical trials. Oncogene. 2011;30(41):4191-4201.
- Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13(5):839-849.
- Jameson JL, Longo DL. Rare diseases and orphan drugs: a potential role for medical genetics. N Engl J Med. 2015;372(17):1646-1654.
附件列表
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。